
Orphan drug designation (ODD) is granted to drugs that are intended to treat rare diseases. This designation provides various incentives — such as tax credits, reduced fees and extended market exclusivity — to encourage pharmaceutical companies to invest in the development of treatments for conditions with limited commercial potential.
Despite the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) offering similar incentives, their processes and requirements for ODD differ significantly. Understanding these differences is crucial for pharmaceutical companies navigating the global landscape of orphan drug development.
So, how do the orphan drug designation criteria differ between the FDA and EMA, and what are the features and benefits of each system?
FDA orphan drug designation
The FDA's orphan drug designation is granted to drugs for rare diseases that affect fewer than 200,000 people in the US or diseases affecting more than 200,000 people where there's no reasonable expectation that the drug will recover the costs of development and marketing.
To apply, you must submit a formal request to the FDA, including detailed information about the rare disease, the drug's mechanism of action and the drug's clinical trial status. Scientific evidence must show the drug has the potential to treat the disease or condition.
Navigating the FDA's requirements for ODD can be challenging for pharmaceutical companies due to the complexities of drug development for rare diseases. However, the FDA's application process is relatively flexible, allowing you to submit a request as early as the pre-clinical phases, which facilitates the drug development process. While ongoing clinical trials can be included in the application, the drug doesn't need to have been fully developed or proven effective at the time of submission.
The FDA may grant accelerated approval for orphan drugs if they meet certain criteria, fast-tracking the drug to market once early-stage evidence indicates efficacy.
The main benefits of the FDA orphan drug designation are:
- Tax credits. The FDA grants up to 25% tax credits on clinical trial expenses related to orphan drug development.
- Exclusivity. Drugs granted orphan drug designation are given seven years of market exclusivity upon approval, meaning no similar drug can be marketed for the same condition during this time.
- Fee reductions. The FDA waives certain application fees, including those associated with new drug applications.
EMA orphan drug designation
The EMA's orphan drug designation applies to drugs intended to treat rare diseases that affect fewer than 5 in 10,000 people in the EU.
The EMA's process for granting ODD differs from that of the FDA, with a stronger focus on scientific advice and the early involvement of regulators.
Like the FDA, you must submit an application detailing the scientific rationale behind the drug's ability to treat a rare disease. You must also provide scientific evidence that the drug is likely to meet the needs of patients, that no other treatments currently exist or that the new drug is a significant improvement.
However, unlike the FDA, the EMA requires a pre-submission meeting for applicants, which helps clarify regulatory expectations and align your application with the agency's standards.
The EMA also uses the Committee for Orphan Medicinal Products (COMP), a specialised body that evaluates applications for orphan drug designation, which ensures a thorough scientific review. Additionally, the EMA offers early involvement in drug development, allowing you to seek advice from the COMP and reduce the risk of misaligned expectations between your company and the regulator.
The primary benefits of the EMA orphan drug designation are:
- Incentives for development. The EMA provides similar incentives to the FDA, including 10 years of market exclusivity once the drug is approved, and fee reductions for regulatory services like the cost of submitting a marketing authorisation application (MAA).
- Scientific support. You can access scientific advice from the EMA early in the drug development process, which can increase the likelihood of regulatory approval.
- Access to EU-wide market. Once approved, your orphan drug can be marketed across all EU member states, offering broader access to an otherwise limited patient population.
Key differences between these orphan drug designations
Both the FDA and EMA provide vital support for orphan drug development. Understanding the differences between these orphan drug designation processes will help you optimise your strategy.
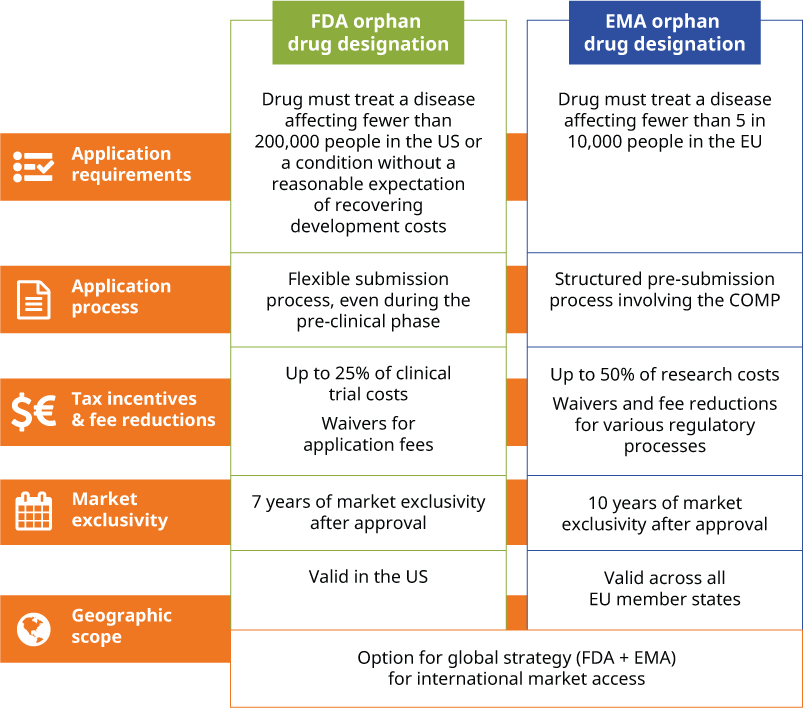
The choice between applying for FDA or EMA orphan drug designation largely depends on your target market, the geographic scope of the product and the drug's development stage. If you have a global strategy, you may choose to pursue an ODD with both agencies, allowing you to leverage the benefits of exclusivity and incentives in the US and Europe simultaneously.
You can significantly improve your chances of securing orphan drug designations and gaining market authorisation approval with TMC's regulatory submission services.
Our expert team can help you obtain the necessary scientific evidence to support your drug's development and guide you through the application process, including pre-submission meetings in the EU. Once approved, we can also act as your marketing authorisation holder (MAH) to ensure compliance with all local regulations.
Contact TMC Consulting today at connect@tmcpharma.com to see how we can help you gain ODD status with the FDA or EMA and support your regulatory submissions.