
The path to regulatory approval for rare disease treatments is often a complex and challenging journey. If you're a small to mid-sized biotech or pharma company looking to design and conduct effective rare disease clinical trials, understanding the nuances of different types of regulatory submissions is crucial to achieving success. In rare disease trials, where patient populations are small, the need for accurate, specialised regulatory submission services becomes even more critical.
The rare disease population often present unique challenges in clinical trials. These conditions can span genetic disorders, rare cancers, autoimmune diseases and metabolic diseases, to name just a few.
Cumulatively, rare diseases are not so rare at all. According to The Lancet Global Health, there are approximately 300 million people living with rare diseases to date and 7,000 diagnosable rare conditions worldwide. Yet, only a small fraction of rare diseases (around 5%) have a regulatory-approved treatment.
This limited therapeutic landscape makes the regulatory submission process for rare disease clinical trials a high-stakes endeavour for any biotech/pharmaceutical company seeking to bring a rare drug to the market. With smaller patient populations, there's a need for expert knowledge of rare disease guidelines and tailored strategies.
The challenges of regulatory submissions for rare disease trials
Rare disease trials face unique challenges in the regulatory environment. Regulatory agencies such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have specific guidelines for rare diseases that require specialised knowledge. These guidelines are constantly evolving, and staying on top of these changes is critical for timely and successful regulatory submissions.
In rare disease trials, recruiting enough participants to generate statistically significant data is difficult. This leads to the need for adaptive trial designs and flexible regulatory approaches. However, presenting limited data without proper regulatory guidance can delay or even derail submission approval.
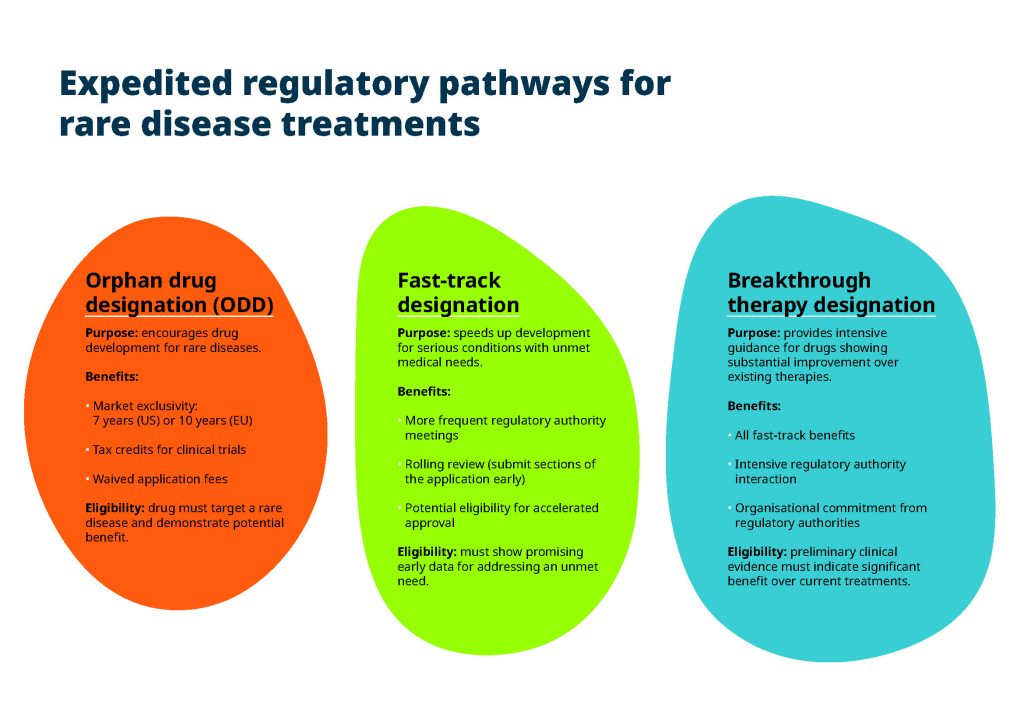
Many rare disease treatments qualify for expedited regulatory pathways like fast track, breakthrough therapy or orphan drug designation. These pathways offer the potential for quicker approval, but they also come with specific requirements and stipulations that can be challenging to navigate without specialised expertise.
The stakes are especially high in rare disease trials, where a failed regulatory submission can result in years of research and development down the drain. The emotional and financial toll on patients and the biopharma companies involved can be devastating. Therefore, an expert hand guiding the regulatory submission process is invaluable.
The benefits of partnering with a rare disease specialist for regulatory submissions
A pharma consultancy company with specialised knowledge of rare diseases understands the intricacies of regulatory frameworks. From orphan drug designation to adaptive trial designs (such as decentralised clinical trial elements), these experts are familiar with the specific regulations that apply to rare disease treatments. This ensures every regulatory submission meets the exact requirements and avoids costly delays.
Regulatory agencies are increasingly open to innovative approaches for clinical trials with small populations, but it takes an experienced pharma regulatory consultant to design and execute the right strategy. A specialist understands how to present limited data, utilise surrogate endpoints and implement other tools that can strengthen the regulatory submission.
Equally, companies working on rare diseases often seek accelerated approval pathways to bring life-saving treatments to patients faster. Specialists in regulatory submissions can help ensure you meet the requirements for expedited programmes like orphan drug designation. Their expertise in these areas can significantly speed up the approval process.
Successful regulatory submissions also require strong relationships with regulatory agencies and a deep understanding of their expectations. A pharma consulting company with regulatory expertise knows how to communicate effectively with the FDA, EMA and other agencies, ensuring that any issues or questions are addressed quickly to keep the regulatory submission process on track.
Accelerate your progress through the regulatory pathway
A specialised pharma services company offers end-to-end support throughout the clinical trial life cycle. From study design and regulatory submissions to clinical trial management and final approval, having a partner with rare disease expertise ensures smooth operations. This holistic approach minimises delays, optimises resources and ultimately leads to successful submission outcomes.
When it comes to rare disease clinical trials, every regulatory submission counts.
By partnering with a specialist pharma consultancy like TMC, you gain access to the expertise necessary to navigate the complex, ever-evolving landscape of regulatory approval. From handling small patient populations to taking full advantage of expedited pathways like FDA or EMA orphan drug designation, working with a specialist ensures your clinical trial submissions are robust, compliant and optimised for success.
Contact TMC Consulting today at connect@tmcpharma.com to accelerate your progress through the regulatory pathway and secure new drug approvals — efficiently and cost-effectively.